Un equipo de investigadores del St. Jude Children´s Research Hospital junto con otros de la Clínica Mayo de Estados Unidos, han identificado un mecanismo biológico que mata las neuronas en la ELA (Esclerosis Lateral Amiotrófica) también llamada enfermedad de Lou Gehring y también en un trastorno genético de demencia frontotemporal (DFT).
El trabajo ha sido publicado en la revista Neuron y ha estado dirigido por J. Paul Taylor, presidente del Departamento de Biología Molecular y Celular de St. Jude e investigador del Instituto Médico Howard Hughes y Rose Rademakers de la Clínica Mayo en Jacksonville, Florida.
Se ha descubierto que la mutación causante de la enfermedad identificada es la primera de su tipo. A diferencia de otras patologías genéticas, la mutación no paraliza una enzima en una vía de regulación biológica; sino que, más bien, la mutación produce una versión anormal de una proteína implicada en un proceso llamado separación de fases en las células.
La separación de fases es un mecanismo por el cual las proteínas se agrupan en conjuntos organizados, llamados organelos sin membrana, necesarios para las funciones celulares ordenadas. Los científicos encontraron que la mutación ELA/DFT produce una versión anormal de una proteína llamada TIA1 que es un componente fundamental de tales organelos.
Como resultado, en la ELA, las proteínas dentro de los organelos se acumulan y matan las neuronas que controlan los músculos. En la DFT, la acumulación mata neuronas en el cerebro. Los investigadores observaron que la separación de fase anormal también puede ser la causa de la enfermedad de Alzheimer.
No existe tratamiento eficaz para la ELA/DFT pero se considera que este descubrimiento ofrecerá una vía para desarrollar tratamientos para restaurar la capacidad de las neuronas de desmontar los organelos cuando su propósito celular ha terminado.
«Estos hallazgos forman parte de un tema emergente de que existe un amplio espectro de enfermedades que incluye la ELA y algunas formas de demencia y miopatía, causadas por alteraciones en el comportamiento de estas estructuras que perturban la organización celular», señala Taylor.
La mutación TIA1 se descubrió cuando los científicos analizaron los genoma de una familia afectada con ELA/DFT. Tras la investigación el efecto de la mutación en la estructura TIA1 los autores descubrieron que se alteraban las propiedades de una cola móvil de la proteína. En esta región de la cola, que regula la capacidad de la proteína para ensamblar con otras proteínas TIA1, Taylor y sus colegas, identificaron las regiones de proteínas no estructuradas llamadas dominios de tipo PRIÓN, como bloques de construcción de ensamblajes celulares y como puntos calientes para las mutaciones causantes de esas enfermedades.
En pacientes con ELA esas mutaciones sucedían con frecuencia al ser analizado el tejido cerebral con mutaciones de pacientes fallecidos. Estos tenían una acumulación de organelos que contenían TIA1 llamados GRÁNULOS DE ESTRÉS en las neuronas.
Los gránulos se forman cuando una célula experimenta tensiones como calor, exposición química y envejecimiento.la célula se aísla en el material genético de los gránulos que codifica proteínas celulares no necesarias para los procesos críticos de supervivencia. Los gránulos también contenían una proteína llamada TDP-43, otro bloque de construcción de gránulos de estrés, cuya anomalía ha sido implicada en la producción de ELA.
En investigaciones con tubos de ensayo y experimentos con células, los científicos detectaron que la mutación TIA1 hace que la proteína se vuelva más pegajosa, retrasando el desensamble normal de los gránulos de estrés, atrapando TDP-43.
Este es el comienzo de un futuro tratamiento efectivo para la ELA y la DFT. Los fármacos no son eficaces actualmente y con esta investigación podría prevenirse el daño neuronal al restaurara el equilibrio saludable de la separación de fases en las células de las personas con mutaciones de las dos enfermedades comentadas.
«Sabemos que estas propiedades materiales están sujetas a una regulación estricta, por lo que quizás no tengamos que atacar la mutación causante de la enfermedad en sí misma comenta Taylor.
Quizás podamos restablecer el equilibrio apuntando a una gran cantidad de moléculas reguladoras en la célula; ya hay enfoques terapéuticos en las pruebas de laboratorio que intentan hacer precisamente eso».
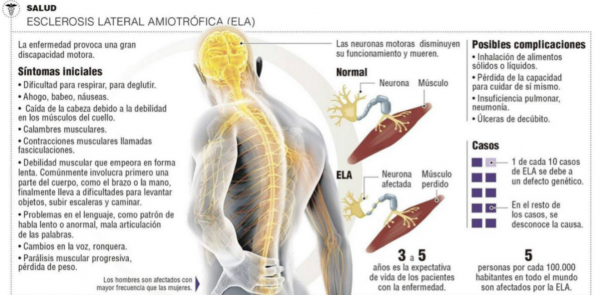
Esclerosis Lateral Amiotrófica (ELA) es una enfermedad neurodegenerativa de tipo neuromuscular. Se origina cuando las células del sistema nervioso llamadas motoneuronas disminuyen gradualmente su funcionamiento y mueren, provocando una parálisis muscular progresiva de pronóstico mortal: en sus etapas avanzadas los pacientes sufren una parálisis total que se acompaña de una exaltación de los reflejos tendinosos (resultado de la pérdida de los controles musculares inhibitorios.


