El síndrome Saethre-Chotzen(SCS), también llamado acrocefalosindactilia tipo III, fue descrito por primera vez por el neurólogo noruego Haakon Saethreen 1931 y el psiquiatra alemán Fritz Chotzenen 1932. Esta enfermedad constituye una craneosinostosis de origen genético que se transmite de padres a hijos según un patrón autosómico dominante.
Las craneosinostosis consisten en el cierre prematuro parcial o total de una o más suturas craneales, con una incidencia de 1 de cada 1000 a 3000 nacidos vivos, con mayor prevalencia en varones. Dentro del contexto de las craneosinostosis primaria, la escafocefalia o craneosinostosis sagital es la forma más frecuente y conocida. Al presentarse en un período de vida donde el crecimiento cráneo cerebral es muy importante, ocasiona en los niños una deformidad craneal característica que debe ser corregida, ya que en caso contrario y al ser progresiva, la afectación estética puede ser muy indeseable; las alteraciones funcionales que se pueden presentar van desde aumento de la presión intracraneana, hasta convulsiones, entre otras.
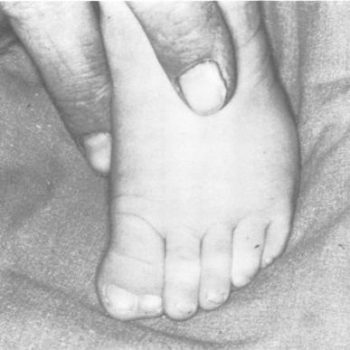
Los niños afectados presentan un espectro variable de manifestaciones desde las primeras horas de vida tales como sinostosis de las suturas coronarles, y en alguna ocasión sagital o lambdoideas lo que produce una forma anómala en el cráneo. Tienen una línea de la frente baja, sufren de estrabismo y tienen una asimetría facial que claramente los identifica además de tener los dedos de los pies anchos y alguna falange distan duplicada en ocasiones.
La inteligencia de estos niños es absolutamente normal en la mayoría de los casos aunque en algunos individuos con grandes delaciones es frecuente que puedan existir retrasos en el desarrollo. La craneosisnostosis tiene lugar cuando existen mutaciones o delaciones puntuales que afectan al gen TWIST1 que es el que codifica un factor de transcripción hélice-bucle-hélice básico responsable de la determinación y la diferenciación de la citocinesis.
Las mutaciones de pérdida de actividad en este gen provocan la inducción de fusiones prematuras de las suturas craneales. Las mutaciones se detectan en cerca del 80% de los individuos afectados mediante una combinación de análisis de deleción/duplicación y análisis de secuencia.
Mediante un análisis genético en la secuencia del exón 1 del gen TWIST1 (único que se traduce) se detectan todas las mutaciones intragénicas en este gen.
Estas deleciones génicas pueden causar fenotipos más graves, normalmente asociados a retrasos neurocognitivos significativos. Se han comunicado mutaciones en FGFR3, FGFR2 y TCF12 que producen síndromes de craneosinostosis que se solapan fenotípicamente con el SCS.
La prevalencia de esta enfermedad genética es de uno cada 25.000 a 50.000 personas aunque existen muchos pacientes que no están diagnosticados debido a que la frecuencia de los síntomas son muy leves y pasan inadvertidos en muchos niños.
La craneosinostosis se presenta con diferentes cuadros clínicos, dependiendo de la extensión y número de suturas fusionadas. Estos van desde alteraciones estéticas a los síntomas funcionales, tales como aumento de la presión craneal, hidrocefalia, déficit visual y trastornos neuropsiquiátricos.
Estas alteraciones son los principales problemas funcionales asociados con craneosinostosis sindrómicas. El aumento de la presión intracraneal se encuentra más comúnmente en pacientes con sinostosis de suturas múltiples. Existen datos recientes que utilizan la grabación prolongada de la presión intracraneal y sugieren que el aumento de esta también puede ocurrir en un paciente con una sola sutura fusionada de manera ocasional.
El aumento de la presión intracraneal parece ser de baja calidad, intermitente y crónico; la frecuencia es mayor en los primeros dos años de vida, y la presión tiende a normalizar después de seis años de edad. Debido a que la presión es de bajo grado, los síntomas clínicos son sutiles o ausente en la mayoría de los casos; una presión cerebral superior a 15 mmHg se define como hipertensión intracraneal acompañado de síntomas clínicos que incluyen dolor de cabeza, irritabilidad y dificultad para conciliar el sueño.
La cefalea es el síntoma clásico asociado con un aumento de la presión intracraneal por cualquier causa; sin embargo, los niños con craneosinostosis y aumento de la presión intracraneal parecen experimentar dolor de cabeza de manera inconsistente.
Otros trastornos funcionales asociados con craneosinostosis son las anormalidades de los nervios craneales pero éstos son relativamente poco comunes, los nervios craneales afectados con mayor frecuencia son I, II, V, VI y VIII. Los síntomas y signos son anosmia, disminución de la agudeza visual, la ceguera, la alteración de la sensibilidad de la cara, neuralgia del trigémino, estrabismo medial, la pérdida de función del nervio craneal afectada, tinnitus audición y vértigo. La epilepsia parece ser más común cuando las múltiples suturas están involucradas y los tipos reportados varían ampliamente y parecen ser el resultado de la misma encefalopatía difusa como retraso mental.
La tomografía computarizada (TC) es el método estándar para investigar una potencial craneosinostosis, y se han propuesto las imágenes 3D; las tomografías computarizadas permiten excelentes imágenes de alta definición de la estructura ósea subyacente, y esto proporciona una guía invaluable como herramienta de diagnóstico para reconocer el tipo de anomalías y en la planificación preoperatoria de la corrección quirúrgica.
La tasa de crecimiento de los picos de cerebro durante el primer año de vida y en la ausencia de la corrección quirúrgica, el cráneo sinostótico progresa a deformidades crecientes que involucran la bóveda craneal, la base del cráneo y el esqueleto facial. La cirugía se realiza en la primera infancia, porque la bóveda craneal en un niño de 3-9 meses sigue siendo fácil de moldear.
El tratamiento de las craneosinostosis es eminentemente quirúrgico y este debe realizarse tempranamente, antes de los 12 meses de edad, el objetivo es descomprimir el cráneo y remodelarlo, disminuir la presión endocraneana, prevenir problemas visuales y del desarrollo mental. En los casos severos requieren de cirugía en los primeros días o semanas de nacidos.


